Introduction to RMVar
RNA modifications can functionally modulates mRNA metabolism and affect diverse eukaryotic biological processes. The correct deposition of RNA modifications is required for normal development. Recent studies have revealed that RNA modification variants are closely related to the dysregulation in cellular processes, leading to serious diseases such as cancer. Functional variants, especially cancer mutations, can significantly alter the status of RNA modifications, leading to the gain or loss of RNA modification sites. RMVar is specifically designed to collect such functional variants, and aimed at providing potential help for revealing the functional roles of RNA modification variants. RMVar contains several most common RNA modifications, including N6-methyladenosine (m6A), N6-dimethyladenosine (m6Am), N1-methyladenosine (m1A), pseudouridine (ψ), 5-methylcytosine (m5C), ribose methylations (2′-O-Me) and 7-Methylguanosine (m7G), 5-methyluridine (m5U), Adenosine-to-inosine (A-to-I). So far, 1,457,898 germline mutations from dbSNP, HGVD, and 220,228 somatic mutations from TCGA, ICGC and COSMIC has been included in RMVar. The experimental evidence of RBP-binding regions and miRNA-RNA interactions as well as splicing sites are also involved in RMVar. In addition, to uncover the underlying relationship between RNA modification machinery and diseases, RMVar establishes an integrated resources combining disease-associated data from GWAS and ClinVar database. Furthermore, multiple statistical diagrams and genome browser are also embedded in the web server for visualizing the analysis results. Currently, users can query or browse the following information from RMVar:
RNA modification associated genetic mutations (dbSNP and HGVD)
RNA modification associated cancer somatic mutations (TCGA, ICGC and COSMIC)
RNA binding protein affected by RNA modification associated variants
miRNA targeting and processing affected by RNA modification associated variants
Splicing sites affected by RNA modification associated variants
Disease related RNA modification associated variants (GWAS and ClinVar)
circRNA related RNA modification associated variants
Data sources and used tools
Pipeline Construction
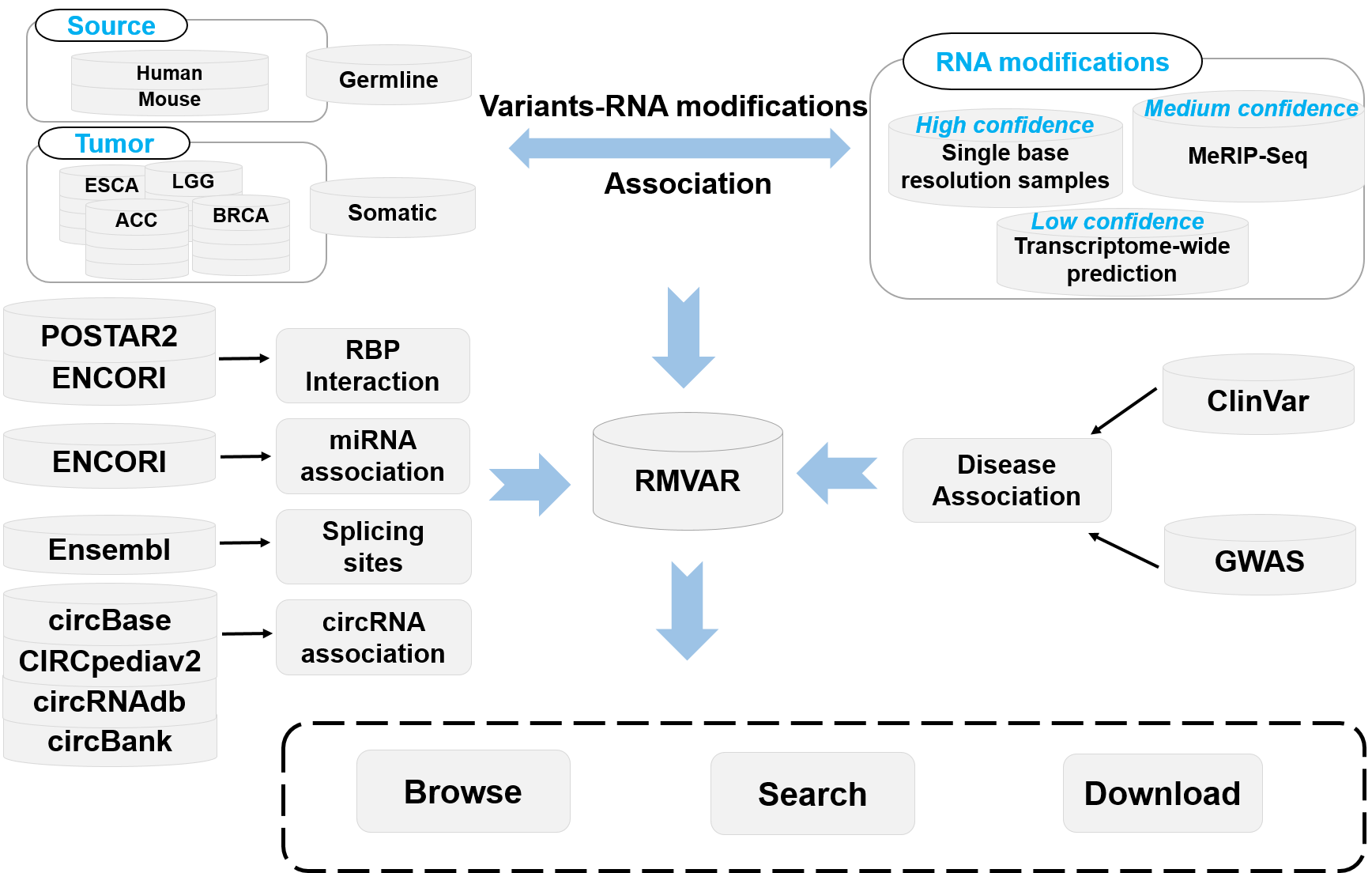
1) Collections of RNA modification data
We manually collected 28 miCLIP samples(Linder et al., 2015 and Moore et al., 2014) 2 PA-m6A-Seq experiments, 2 m6ACE-Seq experiments, 3 DART-Seq experiments, 7 m6A-REF-Seq experiments, 4 MAZTER-seq experiments, 2 m1A-quant-seq experiments, 2 m1A-IP-Seq experiments 2 m1A-MAP experiments, 5 m7G-seq, 10 BS-Seq experiments, 2 Nm-Seq experiments, 14 RiboMeth-Seq experiments, 2 ψ-Seq experiments, 1 DM-ψ-Seq experiment, 3 Ceu-Seq experiments, 3 RBS-Seq experiments and 507 MeRIP-Seq samples from GEO database. All raw sequencing data were downloaded and mapped to human (version: hg38) or mouse (version: mm10) genome. For MeRIP-Seq data, we applied MACS2, MeTPeak for peak calling. In order to ensure a high veracity of data, MSPC was used to combine results and construct consensus peaks from the above three methods.
2) RNA modification associated variants
We downloaded the germline mutations of human and mouse genome from dbSNP and HGVD, and the somatic mutations from TCGA, ICGC and COSMIC. The functional impact of genetic variants on m6A modification is evaluated based on the destruction of conventional DRACH motifs and sequence features. To identify RNA modification loss mutations, we extracted RNA modification sites from the single base resolution experiment samples and overlapped with all mutations to find the functional variants which destroyed the modificaton motifs. Besides, for those mutations that located in consensus peaks from MeRIP-Seq samples, we predicted the functional loss variants that potentially change m6A motifs. In addition, a genome-wide prediction was performed to obtain the potential functional variants based on Random Forest algorithm.
3) RNA binding protein affected by RNA modification associated variants
All RBP binding regions were extracted from ENCORI and POSTAR2, then intersected with the RNA modification associated variants to identify the RBP binding regions affected by RNA modification variants
4) miRNA targeting and processing affected by RNA modification associated variants
Mutations may destroy or create miRNA binding sites on RNA. We therefore downloaded all miRNA-RNA interaction regions from ENCORI, then intersected it with RNA modification associated variants to reveal potential impact on miRNA-mRNA interactions of functional variants.
5) Splicing sites affected by RNA modification associated variants
It is reported that RNA modifications can regulate alternative splicing. We extracted 100 base pairs (bp) upstream form 5' splicing sites and 100bp downstream from 3' splicing sites at all canonical splicing sites (GT-AG), and excluded intron splicing sites with less than 100bp as well as pseudogenes. After that, all m6A-associated variants intersected with these regions to obtain those splicing events affected by RNA modification associated variants.
6) Disease related RNA modification associated variants (GWAS and ClinVar)
GWAS tagSNPs were downloaded from NHGRI GWAS Catalog, Johnson and O' Donnell, dbGAP, GAD, denovo-db, ESP, DisGeNet, GWASATLAS and GRASP database. Haploview was installed to perform LD analysis. Then, for each tagSNP, we use pLink to obtain its LD mutations in different populations (The used populations include CHB, CEU, JPT, TSI). Moreover, we also collected ClinVar data to provide further information about the relationship between RNA modification associated alterations and disease.
7) circRNA related RNA modification associated variants
Accumulation of circRNAs could be controlled by RNA modifications. We therefore downloaded all circRNA regions from circBase, CIRCpediav2, circRNAdb and circBank, then intersected it with RNA modification associated variants to reveal potential relationship between RNA modifications and circRNAs.
The definition of gain and loss RNA modification associated mutation
In order to predict RNA modification associated variants, we utilize a deep convolutional neural network model to predict and annotate the functional modification sites. Combining all variants in the prediction model, we separately predicted the RNA modification status in reference sample and mutant sample. We defined an RNA modification gain mutation as the RNA modification was occurred in mutated RNA sequence but not in reference sequence. While an RNA modification loss alteration would be defined as the opposite case.
Search in RMVar
For convenience, we provide a user-friendly web interface for RMVar database. Users can browse or search the data at different levels.
Browse:
1) Browse by different species in Germline
2) Browse by different tumors in TCGA&ICGC
3) Browse by different tissues in COSMIC
Quick Search:
1) Quick search for RMVar ID
2) Quick search for RsID of SNP
3) Quick search for genes
4) Quick search for an interested region
5) Quick search for an interested disease
Advance Search:
Multiple conditions can be easily combined, and you can obtain your specific queries.